Research Directions
We develop theoretical and computational methods to understand quantum phenomena in chemical and biological systems, from electronic structure to reaction dynamics.
Multi-State Density Functional Theory (MSDFT)
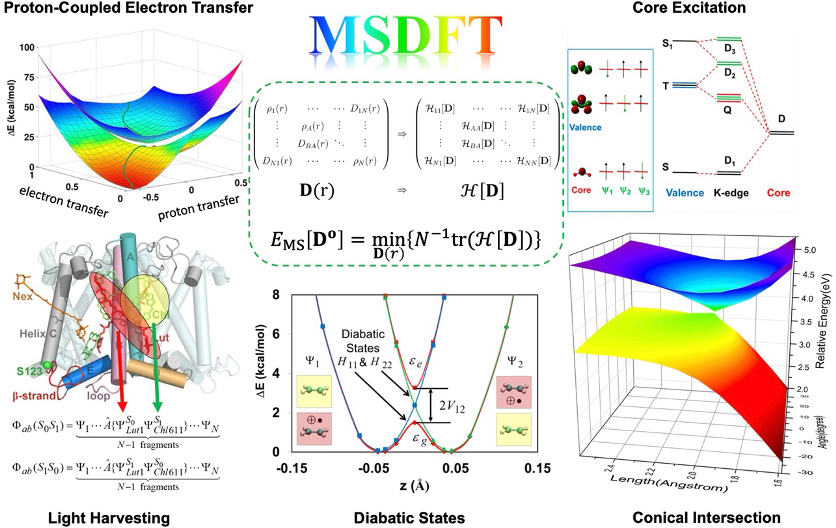
Current electronic structure theory faces significant challenges in achieving a balanced description of both dynamic and static correlation effects, especially for multiple electronic states. While Kohn-Sham Density Functional Theory (KS-DFT) and its time-dependent extension (TDDFT) are remarkably successful for systems dominated by a single reference configuration, they systematically fail for strongly correlated systems, charge-transfer excitations, and double excitations. On the other hand, multi-reference wavefunction methods, though in principle exact, are plagued by unfavorable computational scaling, limiting their application to small molecules.
To address these fundamental limitations, Multistate Density Functional Theory (MSDFT) provides a rigorous, time-independent DFT framework for all electronic states. Its core innovation is the introduction of an N-rank matrix density, D(r), which contains both state densities (diagonal elements) and transition densities (off-diagonal elements), as the fundamental variable. A foundational theorem establishes a one-to-one correspondence between D(r) and the Hamiltonian matrix projected within the subspace of the lowest N eigenstates, defining a Hamiltonian matrix functional, H[D]. A variational principle is derived: minimizing the multistate energy (the trace of H[D]) with respect to a trial D(r) yields, at convergence, the exact energies and densities of the N-lowest eigenstates. Crucially, a theorem proves that any N-matrix density can be exactly represented by no more than N² Slater determinants, defining a minimal active space (MAS). This naturally leads to the definition of a correlation matrix functional, Ec[D], which is the multi-state generalization of the exchange-correlation functional in KS-DFT, capturing electron correlation effects within the subspace.
For practical computations, the Non-Orthogonal State Interaction (NOSI) algorithm has been developed. It involves independently optimizing the Slater determinants within the MAS using occupation-constrained methods like Block-Localized Excitation (BLE) or Target State Optimization (TSO) to prevent variational collapse. Approximations are then introduced for the correlation matrix functional: the diagonal elements are approximated using standard KS-DFT energy functionals, while off-diagonal elements, particularly for spin-coupling, are determined by enforcing spin-multiplet degeneracy constraints or an overlap-weighted average of state correlation energies. The method has demonstrated considerable successes in applications where conventional methods struggle. This includes providing accurate vertical excitation energies for small molecules, correctly describing charge-transfer excitations in donor-acceptor complexes, quantitative description of avoided crossings and conical intersections, and computing core-level excitation spectra for both closed-shell molecules and open-shell radicals—all with an accuracy rivaling high-level wavefunction theories but at a computational cost comparable to a set of individual KS-DFT calculations.
Quantum Biology
Quantum biology is an interdisciplinary field that investigates the potential role of non-trivial quantum-mechanical phenomena in biological processes. It challenges the classical view of life by exploring how effects such as coherence, spin coupling, and quantum tunneling might be harnessed within the warm, wet, and seemingly noisy environment of living organisms to confer functional advantages. Key areas of inquiry include the potential role of electronic/vibronic coherence in optimizing biological electron/energy transfer reactions, the hypothesized functional role of spin-correlated radical pairs and entanglement in biological magneto-reception, and the influence of nuclear quantum effects (like proton delocalization) on hydrogen bonding and reaction dynamics in proteins. The central goal is to determine whether these quantum effects are incidental or are actively exploited by evolution to enable functions that would be less efficient or impossible with purely classical mechanisms.
Qbics (Quantum, bio-informatics, and chemical sciences) is a general-purpose computational platform for accurate and efficient simulations of biological and chemical phenomena that is independently developed in the Shenzhen Bay Laboratory. Our research group intends to leverage and develop novel theoretical methods, such as MSDFT, and computational algorithms within Qbics to advance the understanding of quantum effects in biology. This includes developing and applying methods for large-scale excited-state simulations, modeling nonadiabatic dynamics, QM/MM simulation of enzyme catalysis, path-integral simulation of nuclear quantum effects for proton transport/tunneling and hydrogen bonds, and modeling spin dynamics of radical pairs in biology, such as magnetoreceptor proteins. By integrating these advanced computational techniques, we seek to provide quantifiable, mechanistic insights that bridge the gap between quantum physics and biological function, for resolving molecular mechanisms and ultimately rational design of biological functions.
1. Ultrafast reaction dynamics of photo-enzymes and photo-receptors
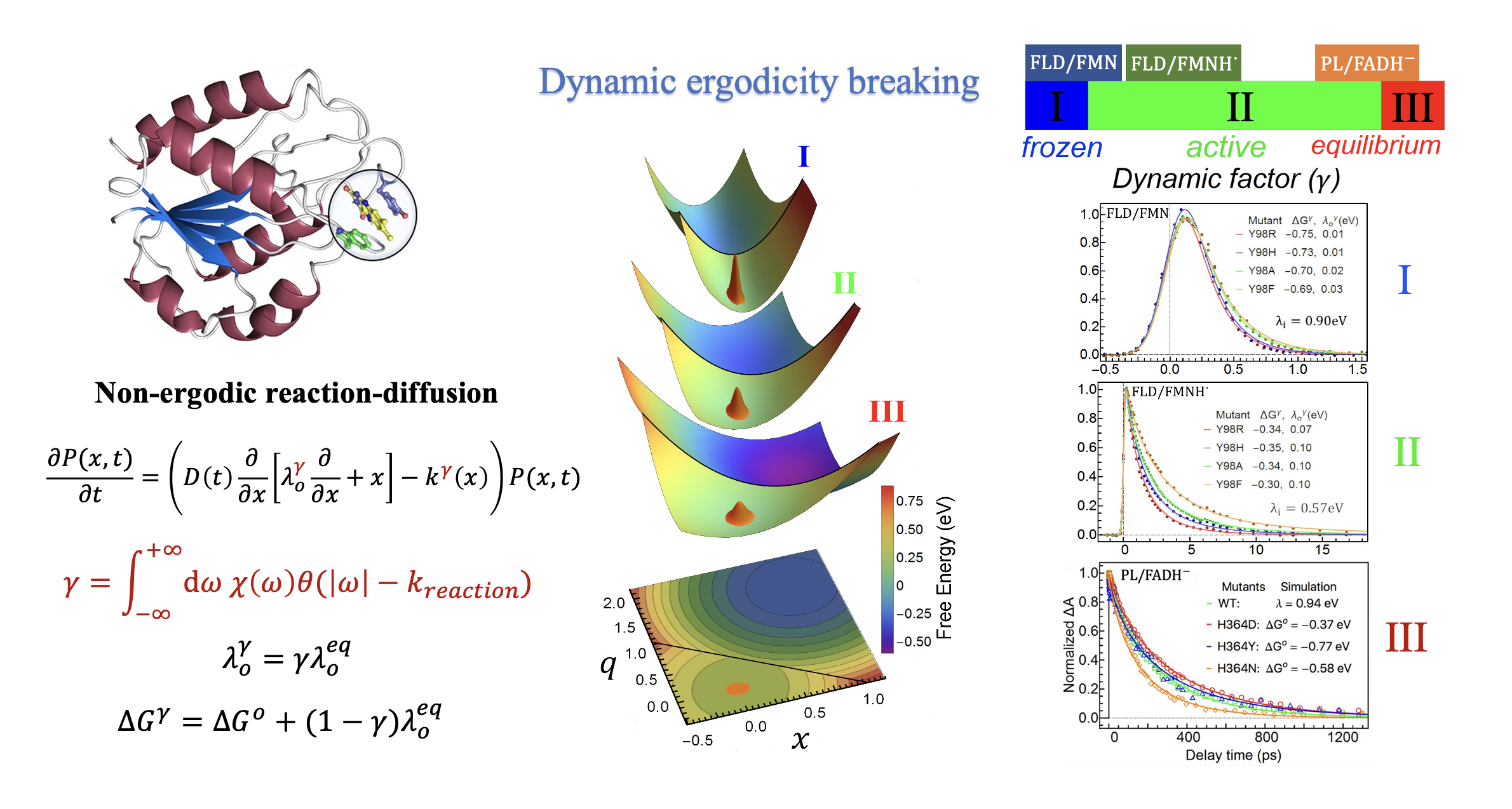
Living systems are intrinsically non-equilibrium, in which dynamical processes occur across multiple spatial and temporal scales. Biological functions are manifested as inter-connected dynamical processes of different scales, making it a grand challenge to unravel their principal mechanisms. As the elementary reactions underlying biological functions, ultrafast reactions in proteins, ranging in timescales from femtoseconds to a few nanoseconds, are immersed in a highly heterogeneous environment with motions spanning over a continuous spectrum of timescales from femtoseconds to microseconds. Because of the multi-scale non-equilibrium environment, as a result of photoexcitation of chromophores embedded in the active site, the electron transfer (ET) dynamics in proteins displays a variety of behaviors, failed to be described by traditional theories. We proposed that the reason for the complex reaction dynamics is due to the fact that the protein's phase space is ergodicity broken. We developed a kinetic theory to describe the non-ergodic reaction dynamics and introduced a unified scheme for categorization of various types of ET dynamics observed in proteins. Since then, this novel phenomenon, known as dynamic ergodicity breaking, has been observed in a number of photo-receptors and photo-enzymes, including flavodoxin and photolyase. Another work of ours reflects a further layer of complexity in reactions of proteins. We developed a theoretical model to describe the ultrafast dynamics of two coupled ET reactions, each of which is further correlated with motions in the local environment. The applicant also developed models to study the role of nuclear quantum effects, i.e., vibrational excited states, in ultrafast reaction dynamics.